
This information is for health professionals and service providers.
Motor neurone disease (MND) is a fatal, progressive, degenerative, neurological condition. It is the name given to a group of diseases in which the nerve cells controlling the muscles that enable us to move, speak, breathe and swallow undergo degeneration and die.
MND is also known as Amyotrophic Lateral Sclerosis (ALS) in other parts of the world.
The causes of the majority of cases of MND remain unknown. However, about 15% of cases are inherited (familial) and the genetic fault of about 60% of these cases is now known in Australian families. However, current research is showing that many of the sporadic cases also have a genetic component.
Average survival time after diagnosis is 2.5 years, but a minority, around 5 to 10%, will survive for more than 10 years. MND affects each person differently in respect of initial symptoms, rate and pattern of progression and survival time.
MND occurs in all countries of the world. The impact of ALS/MND on the community is usually measured by the incidence and prevalence of the disease. Incidence is the number of new cases added in a defined period, usually a year. Prevalence is the number of cases existing at any point in time.
Worldwide the incidence of ALS/MND is 2 per 100,000 of total population, while the prevalence is around 6 per 100,000 of total population. Research has found that the incidence is higher in people aged over 50 years. Although classified as a rare disease based on its prevalence it is estimated that approximately 140,000 new cases are diagnosed worldwide each year. The lifetime risk of developing ALS/MND has been calculated to be 1/300.
In Australia, on average, each day two people die from MND and two people are diagnosed with MND.
A Deloitte Access Economics report commissioned by MND Australia in 2015 estimated that there were 2,094 Australians living with MND, of whom 60% were male and 40% female. The overall prevalence rate was estimated to be 8.7 per 100,000 Australians, or 1 in 11,434 Australians, with the highest prevalence rate reported in males aged between 75 and 84 years.
The report found that in comparison to other countries, Australia has a relatively high level of prevalence, with recent studies conducted in Europe finding prevalence rates of 7.9 per 100,000 population.
Mortality rates due to MND are relatively high in all age groups. The mortality rate is estimated to be 3.14 per 100,000 across the entire population, which was equivalent to 752 deaths due to MND in 2015. Approximately 55% of deaths occur in males. Deaths associated with MND are typically due to respiratory failure as a result of weakened respiratory muscles.
.png "About-MND-(1).png")
Latest Australian Institute of Health and Welfare (AIHW) and Australian Bureau of Statistics (ABS) figures indicate that 862 people died in Australia from, and with, MND in 2019.
The global incidence of ALS varies between 1 and 2.6 cases per 100,000 people per year, with the average age of onset ranging from 54 to 67 years old. The prevalence of ALS increases with age, reaching 1/5000 among people aged 70–79 years old. Consequently, as the population ages, it is expected that the world’s total number of cases will reach more than 375,000 by 2040. Owing to the lack of a reliable diagnostic test, absence of validated biomarkers, and phenotypes that are easily confounded with other MNDs, including primary lateral sclerosis (PLS) and progressive muscular atrophy (PMA), there is a delay of approximately 11–12 months in reaching a definite diagnosis. Currently, diagnosis is based on a set of clinical criteria (El Escorial and revisions, and Awaji-Shima criteria) that can be used to stratify patients according to the area of initial onset and the progression of symptoms (Connolly et al 2020).
The incidence of ALS is about 2 per 100,000 person-years, and the prevalence is about 5 per 100,000 persons. Because of the low prevalence, the average primary care physician will see 1 person in their lifetime, a typical UK neurologist will diagnose about 2 people a year, while a tertiary referral centre will see more than 100 people. Despite the low incidence, however, ALS is not particularly infrequent. The lifetime risk is about 1 in 300 by the age of 85, with the risk increasing steadily, at least until about the eighth decade of life. This is very similar to the risk for multiple sclerosis in the UK (Martin et al 2017).
Contrary to earlier assumptions, the incidence of ALS has been shown to differ based on ancestral origin; studies in populations of European origin have shown a crude incidence of >3 cases per 100,000 individuals but incidence rates are lower in East Asia (around 0.8 per 100,00) and South Asia (0.7 per 100,000).
In some regions (such as Guam and the Kii peninsula of Japan) the reported incidence was very high, but dropped substantially over the past 30 years for reasons that remain unclear. In areas where different ancestral populations live in close proximity (as in Northern America), the incidence rates of ALS in indigenous populations is particularly low (0.63 cases per 100,000 individuals), whereas reported incidences in regions of relatively homogeneous populations (such as Ireland, Scotland and the Faroe Islands) are high (2.6 cases per 100,000 individuals) (Hardiman et al 2017).
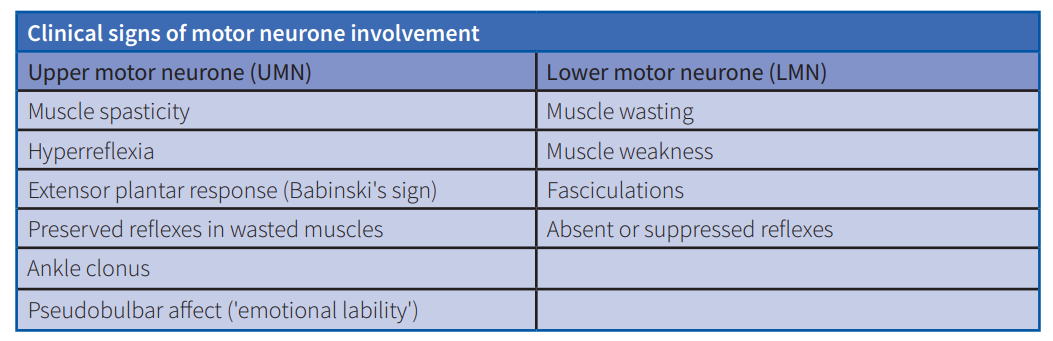
MND onset is insidious. Initial symptoms may be mild at first and can include stumbling due to weakness of the leg muscles, difficulty holding objects due to weakness of the hand muscles, slurring of speech and swallowing difficulties due to weakness of the tongue and throat muscles, or shortness of breath due to respiratory muscle involvement. Some people may present with frontotemporal cognitive changes. Emotional responses may be more easily triggered, and the person with MND may be aware of laughing and crying more readily than previously. Cramps and muscle twitching are also common symptoms.
Average age of onset is most commonly the middle years and onwards.
Typically people present with symptoms in one muscle group, for example weakness and wasting of one hand or a unilateral foot drop. MND can be categorised on the basis of sites of involvement at presentation and the balance between lower motor neurone and upper motor neurone features. The demarcation between the different MND clinical groups is frequently blurred. As the disease progresses there may be considerable overlap resulting in more generalised muscle wasting and weakness.
Sensory symptoms or signs are rare and should lead to review of the diagnosis.
The motor nuclei controlling eye movements and the voluntary pelvic sphincter muscles usually remain intact.
.png "signs-mnd-(1).png")
The majority of ALS cases (~70%) have spinal onset, usually presenting with focal limb weakness such as foot drop or a weak hand. The disease then tends to spread in a contiguous manner, initiating at distinct focal regions of the body and then propagating from the primarily affected area to adjuvant secondary sites of the body.
In 25% of ALS cases, symptoms develop initially in the bulbar-innervated muscles. Bulbar-onset ALS is more common in women, especially after 70 years (M:F ratio 1:1.6). Dysarthria almost always predates dysphagia and cognitive impairment is often present. Approximately 3% to 5% of patients present with respiratory or cognitive onset. Thoracic spinal-onset ALS can present as truncal weakness or respiratory impairment and is associated with poor prognosis, with a mean survival time of just 1.4 years.
Cognitive-onset ALS patients usually present symptoms characteristic of frontotemporal dementia (FTD), such as changes in behaviour, personality and cognition which are all suggestive of frontal impairments.
In summary, initial site of symptom onset varies among ALS patients from classic limb-onset to rare cognitive-onset phenotypes, and a poor prognosis is often associated with bulbar and respiratory onset (Connolly et al 2020).
(ALS) is a progressive, paralytic disorder characterized by degeneration of motor neurons in the brain and spinal cord. It begins insidiously with focal weakness but spreads relentlessly to involve most muscles, including the diaphragm. Typically, death due to respiratory paralysis occurs in 3 to 5 years.
Motor neurons are grouped into upper populations in the motor cortex and lower populations in the brain stem and spinal cord; lower motor neurons innervate muscle. When corticospinal (upper) motor neurons fail, muscle stiffness and spasticity result. When lower motor neurons become affected, they initially show excessive electrical irritability, leading to spontaneous muscle twitching (fasciculations); as they degenerate, they lose synaptic connectivity with their target muscles, which then atrophy.
ALS is the most frequent neurodegenerative disorder of midlife, with an onset in the middle-to-late 50s. An onset in the late teenage or early adult years is usually indicative of familial ALS. The time from the first symptom of ALS to diagnosis is approximately 12 months, a problematic delay if successful therapy requires early intervention. Because an abundance of ALS genes have now been identified, it will probably be informative to re-analyze this epidemiologic profile of ALS with stratification according to genetically defined subtypes (Brown and Al-Chalabi 2017).
Motor neuron disease (MND) (also known as amyotrophic lateral sclerosis (ALS) and more colloquially Lou Gehrig's disease) is a rapidly progressive and universally fatal neurodegenerative disorder of the human motor system, first described in the mid‐19th century. Although MND is clinically characterised by progressive neurological deterioration and coexistence of upper and lower motor neuron signs, recent discoveries have indicated a heterogeneous nature of MND. Despite the clinical heterogeneity, median survival of MND remains 3 years, with 10% of patients surviving over 8 years (Simon et al 2015).
MND can be classified into four main types depending on the pattern of motor neurone involvement and the part of the body where the symptoms begin.
1. Amyotrophic lateral sclerosis (ALS)
- both upper and lower motor neurones are affected
- limb muscle weakness and wasting
ALS is the most common type, characterised by muscle weakness and stiffness, over-active reflexes and, in some cases, rapidly changing emotions. Initially the limbs cease to work properly. The muscles of speech, swallowing and breathing are usually also later affected. ALS is the term commonly applied to MND in many parts of the world.
2. Progressive bulbar palsy (PBP)
- both upper and lower motor neurones are affected
- speech and swallowing muscle weakness and wasting
When ALS begins in the muscles of speech and swallowing it is designated progressive bulbar palsy. Progressive bulbar palsy (PBP), also called progressive bulbar atrophy, involves the brain stem – the bulb shaped region containing lower motor neurones needed for swallowing, speaking and chewing. Symptoms include pharyngeal muscle weakness (involved with swallowing), weak jaw and facial muscles, progressive loss of speech and tongue muscle atrophy. Limb weakness with both lower and upper motor neurone signs is almost always evident but less prominent.
Pseudobulbar palsy shares many of the symptoms of progressive bulbar palsy (PBP) and is characterised by degeneration of upper motor neurones that transmit signals to the lower motor neurones in the brain stem. Affected individuals have progressive loss of the ability to speak, chew and swallow. Progressive weakness in facial muscles leads to an expressionless face. Individuals may develop a gravelly voice and an increased gag reflex. People with PBP or pseudobulbar palsy may have outbursts of emotional lability eg; laughing or crying uncontrollably.
3. Progressive muscular atrophy (PMA)
- lower motor neurones are affected
- slower rates of progression and significantly longer survival compared to ALS and PBP
PMA is characterised initially by lower motor neurone signs resulting in more generalised muscle wasting and weakness, absent reflexes, loss of weight and muscle twitching. PMA can be the hardest form of MND to diagnose accurately. Weakness is typically seen first in the hands and then spreads into the lower body where it can be severe. Other symptoms may include muscle wasting, clumsy hand movements, fasciculations and muscle cramps. The trunk muscles and respiration may be affected. Recent studies indicate that many people diagnosed with PMA subsequently develop upper motor neurone signs. This would lead to a reclassification to ALS. PMA may begin in the arms (flail arm type) or the legs (flail leg type).
4. Primary lateral sclerosis (PLS)
- upper motor neurones are affected
- very rare and diagnosis is often provisional
PLS affects the upper motor neurones of the arms, legs and face. It occurs when specific nerve cells in the motor regions of the cerebral cortex gradually degenerate, causing the movements to be slow and effortful. The disorder often affects the legs first, followed by the body, trunk, arms and hands and, finally, the bulbar muscles. Speech may become slowed and slurred. When affected, the legs and arms become stiff, clumsy, slow and weak, leading to an inability to walk or carry out tasks requiring fine hand coordination. Difficulty with balance may lead to falls. Affected individuals commonly experience pseudobulbar affect and an overactive startle response.
MND/FTD
A small proportion (5–15%) of people with MND will receive a diagnosis of ‘motor neurone disease with fronto-temporal dementia’ or MND/ FTD. Often the symptoms of dementia precede the motor symptoms, sometimes by a number of years.
5 major clinical phenotypes were determined and included ALS bulbar onset, ALS cervical onset and ALS lumbar onset, flail arm and leg and primary lateral sclerosis (PLS). Of the 1834 registered patients, 1677 (90%) could be allocated a clinical phenotype. ALS bulbar onset had a significantly lower length of survival when compared with all other clinical phenotypes (p<0.004). There were delays in the median time to diagnosis of up to 12 months for the ALS phenotypes, 18 months for the flail limb phenotypes and 19 months for PLS (Talman et al 2016).
....with profound clinical, prognostic, neuropathological, and now genetic heterogeneity, the concept of ALS as one disease appears increasingly untenable. This background calls for the development of a more sophisticated taxonomy, and an appreciation of ALS as the breakdown of a wider network rather than a discrete vulnerable population of specialised motor neurons (Turner et al 2013).
Amyotrophic lateral sclerosis (ALS) is characterized phenotypically by progressive weakness and neuropathologically by loss of motor neurons. Phenotypically, there is marked heterogeneity. Typical ALS has mixed upper motor neuron (UMN) and lower motor neuron (LMN) involvement. Primary lateral sclerosis has predominant UMN involvement. Progressive muscular atrophy has predominant LMN involvement. Bulbar and limb ALS have predominant regional involvement. Frontotemporal dementia has significant cognitive and behavioral involvement. These phenotypes can be so distinctive that they would seem to have differing biology. However, they cannot be distinguished, at least neuropathologically or genetically (Ravits et al 2013).
Kennedy’s Disease
Kennedy’s disease (KD), also known as X-linked spinobulbar muscular atrophy (SBMA), is an inherited disorder of the lower motor neurones. Due to similar symptoms, people with KD are sometimes misdiagnosed as having MND. Kennedy's disease is characterised by slowly progressive weakness and wasting of muscles with only lower motor neurone involvement and other features.
- Kennedy's disease is an inherited disorder affecting adult males
- Females who have a father with KD are inevitably KD gene carriers and their sons (and daughters) have a 50% chance of inheriting the KD gene
- Female gene carriers do not usually develop any KD symptoms
- Males who have inherited the gene will develop KD symptoms as adults
- Kennedy's disease cannot be passed from father to son
Clinically the sporadic and familial forms of MND are indistinguishable.
- familial MND accounts for about 10% of all MND cases
- MND is sporadic in about 90% of cases, developing with no clearly identifiable causes
In recent times the pace of gene mutation discovery has accelerated due to advancements in technology and a concerted international collaborative approach with many mutations that affect small cohorts of people with MND now identified. It is now possible to test for the presence of mutations in the SOD1, TDP43, FUS and C9orf72 and other recently identified genes in a person diagnosed with familial MND.
Research is increasing our understanding of the mechanism by which these mutations cause MND and may lead to treatments which can prevent or delay the onset of MND in someone with an MND-related genetic mutation. Clinical trials of therapies targeting SOD1 and C9orf72 mutations are currently underway.
It is becoming apparent that many sporadic MND cases also have underlying genetic mutations. As we discover more MND causing gene mutations in both familial and sporadic forms of MND, and in light of new personalised therapies in development, offering genetic testing to all people diagnosed with MND may become routine practice. This will require careful discussion with the person diagnosed and their family as well as the availability of professional genetic counselling and access to up to date, evidence based information.
The number of clinically actionable results is likely to increase with the anticipated development of new genetic-therapy approaches for ALS. Previous genetic studies of ALS have been largely retrospective and were therefore unable to determine the utility of genetic screening in the clinic. In contrast, the present study strongly suggests that routine genetic testing should be offered in both patients with familial ALS and those with sporadic ALS, at least in our population (Shepheard et al 2021).
It is easy to demonstrate the harmful consequences of the archaic familial/sporadic division. Let us take the most frequent cause of ALS: expansion mutation in the gene C9ORF72. This is present in about 45% of people with a family history, making it by far the most important cause of 'familial' ALS. But this mutation is also present in up to 10% of the much larger number of people with apparently no relevant family history, so it is the most important cause of 'sporadic' ALS as well. In fact, these mutations account for about 14% of all ALS, and in most cases there will be no family history. How can it make sense to treat people differently on the basis of their family history in this context? What matters is the underlying cause. In these cases, we should be using the term C9ORF72-mediated ALS, not familial or sporadic ALS. Whether there is a family history of the disease is irrelevant to the patient, to the family, to the research team and to our understanding of the mechanism.
Now that gene sequencing is cheap and easy, we need to move away from the outdated, unhelpful familial/sporadic dichotomy and instead use terms that describe what is known about the causes of ALS in each person. Only then can we think clearly, identify risk factors, and counsel patients correctly (Al-Chalabi 2017)
We report 60.8% of Australian ALS families in this cohort harbour a known ALS mutation. Hexanucleotide repeat expansions in C9orf72 accounted for 40.6% of families and 2.9% of sporadic patients. We also report ALS families with mutations in SOD1 (13.7%), FUS (2.4%), TARDBP (1.9%), UBQLN2 (.9%), OPTN (.5%), TBK1 (.5%) and CCNF (.5%) (McCann et al 2017).
Five to 10% of ALS is genetically transmitted mainly by way of dominant gene mutations and these numbers increase to as high as 15–20% when known genes are tested in patients who were thought to have sporadic disease. Approximately 60–70% of FALS pedigrees have genes that have now been identified, the main ones being SOD1, TARDBP, FUS, C9ORF72, OPTN, VCP, UBQLN, and PFN1 (reviewed in (65)). Clinical phenotype heterogeneity of FALS is as characteristic and as vast as SALS, and no clinical features easily distinguish one from another. Remarkably, this clinical phenotype heterogeneity is even seen in the same mutation in the same gene in the same kinship, implying phenotype is likely determined by factors other than the molecular cascade it triggers. However, some trends exist. Mutations in SOD1 and FUS tend to cause predominantly LMN syndromes. Mutations in TARDBP tend to begin in the upper extremity and to progress slower than average. Mutations in SOD1, TARDBP and FUS cause mostly motor syndromes and only rarely associated FTD. Mutations in FUS cause a juvenile as well as adult motor neuron disease syndrome. Mutations in C9ORF72 are as likely to cause FTD as ALS, often with psychosis. The ‘A4V mutation in SOD1 ALS is rapid while the ‘D90A’ mutation, unusual in that it is recessive, slow and indolent (Ravits et al 2013).
In most cases of MND the cause is unknown.
Of the 5-10% of people who develop the familial form of MND, approximately 60–70% of genes involved have now been identified. As genetic factors are identified and metabolic pathways become clearer, the potential environmental factors to test may become more obvious.
Our understanding of the genetic basis of MND is increasing rapidly, but the relationship between genetic risk, environmental exposure and phenotype remains largely unknown particularly in sporadic MND.
There are a number of causes that have been proposed to contribute to the development of MND. These include:
- genetic factors
- physical trauma/exercise
- infection by viral agents
- gut microbiomes
- exposure to environmental toxins and chemicals
A number of different mechanisms that can affect the health of motor neurons are thought to contribute to MND:
- protein aggregation
- glutamate toxicity
- mitochondrial dysfunction
- dysfunctional signalling pathways
- free radical damage
- immune mediated damage
- loss of growth factors required to maintain motor neurone survival
It is thought that these may act individually or in combination to cause or contribute to the development of MND. Recent research suggests a six-step process with genes, environment and ageing contributing to the development of MND. Research focussed on environmental causes, such as high intensity exercise and blue green algae, understandably often provoke intense media and community interest. Some studies may report an association, but it is important to note that association is very different from cause.
More research is needed to improve understanding of the impact of environment particularly on people with a genetic predisposition to MND. The causes and disease mechanisms section of the MND Association of England, Wales and Northern Ireland MND Research Blog provides an overview of recent research on potential causes of MND.
In conclusion, the current evidence supports a complex causal relationship between physical exercise and ALS. However, it is clear that, for the majority of individuals, the health benefits of a physically active lifestyle markedly outweigh the risks. The key objective for future research is to understand which individuals are at risk of developing ALS if they exercise excessively and provide appropriate lifestyle counselling. Our work goes some way towards developing this aim and in particular, we propose that C9ORF72 penetrance may be influenced by high levels of physical activity (Julian et al 2021).
Over 150 years have passed since ALS was first reported by Charcot and still the aetiology of the disease remains elusive. Although research is progressing and genetic studies continue to identify novel gene associations, many questions remain surrounding the pathological mechanisms associated with already established mutations, their role in the ALS phenotype, and the as yet undiscovered mechanisms that underlie sporadic onset of disease (Connolly et al 2020).
This study established a linear relationship between log incidence and log age onset in an Australia ALS population, consistent with a multistep process. Identification of these steps will likely be of therapeutic benefit (Vucic et al 2019).
ALS is considered a complex genetic disorder with a Mendelian pattern of inheritance in a proportion of cases, but no discernible family history in the rest. Mathematical models developed using population-based registers have suggested that individuals with ALS are likely to carry a number of at risk variants that interact with environmental factors through a series of at least 6 notional steps leading to disease manifestation. One of these steps is thought to be the genetic risk (from birth), but the interplay of environmental factors that lead to the remaining steps have yet to be defined. In transgenic mice, the genetic background can alter the phenotypic presentation of ALS suggesting that human disease phenotypes could also have a genetic basis, and that genomic and epigenomic “fingerprinting” could permit the clustering of different phenotypic manifestations into discrete underlying causes that are amenable to therapeutic intervention (Hardiman et al 2017).
The apparently homogeneous phenotype of predominantly motor degeneration that is ALS can result from many different causes: genetic, epigenetic, environmental, and internal. Thus, many different pathways converge on the final outcome of upper and lower motor neuron death. Careful analysis of incidence data in European population registers shows that, on average, each pathway comprises six molecular steps. The model explains many otherwise enigmatic features of ALS, such as the increasing risk with age, genetic pleiotropy (the same gene variation can result in different diseases), age-dependent penetrance of disease genes, the difficulty in identifying a single environmental cause, the observation that ALS appears to start in one region and spread, and that it is specific to motor neurons but can affect other cell types. The next challenge is to understand the extent to which the pathways overlap and therefore might be amenable to a common treatment strategy. Although ALS remains a uniformly fatal diagnosis, accelerating advances in our understanding bring the hope that an effective treatment can be found for this devastating disease (Martin et al 2017).
Our findings support the hypothesis that a multistep process requiring just six distinct steps leads to the onset of amyotrophic lateral sclerosis. This provides hope that the identification of the steps could therefore lead to preventive or therapeutic avenues that have not yet been examined. The consistency of our findings across five western European populations and in both sexes suggests that the underlying mechanism is similar, and increases our confidence in the validity of the results. This hypothesis provides an explanation for several intriguing features of amyotrophic lateral sclerosis and means that environmental studies are essential even in people at high risk because of inherited genetic susceptibility (Al-Chalabi et al 2014).
Approved therapies
Currently only two neuroprotective therapies are available both of which have only very modest effects on the course of the disease.
Riluzole is an anti-glutamate medication that appears to block the release of glutamate from neurones. Riluzole is manufactured under the names Rilutek™ and APO-Riluzole. In Australia riluzole is available for eligible people at a subsidised price on the Pharmaceutical Benefits Scheme (PBS) under an authority prescription. Teglutik® (riluzole) a liquid formulation (for ease of swallowing or use via PEG tube) of riluzole, distributed by Seqirus (Australia) Pty Ltd, was listed on the PBS in April 2019 under the same prescribing conditions as Rilutek™ and APO-Riluzole.
Edaravone (radicava) has been shown to provide a limited improvement in survival in a small sub-set of people with ALS/MND. It works by suppressing oxidative stress and is administered via intravenous infusions. Edaravone has been approved in a number of countries but has not been approved by the therapeutics goods administration (TGA) in Australia.
The mainstay of treatment for people living with MND is coordinated multidisciplinary care and timely access to interventions to manage symptoms.
This review indicates that riluzole treatment may extend survival by 6–19 months, which is far greater than the 2- to 3-month survival benefit reported in original pivotal RCTs. Real-world evidence studies provide valuable information concerning characteristics among the broad patient population observed in clinical practice and physician treatment practices outside clinical trials. This information is necessary to guide treatment decisions and for reimbursement and payment decisions. This analysis of population studies of patients with ALS was conducted to determine if the observed survival benefit of riluzole exceeds what was demonstrated in pivotal RCTs (Andrews et al 2020).
No therapy offers a substantial clinical benefit for patients with ALS. The drugs riluzole and edaravone, which have been approved by the Food and Drug Administration for the treatment of ALS, provide a limited improvement in survival. Riluzole acts by suppressing excessive motor neuron firing, and edaravone by suppressing oxidative stress. Numerous other compounds that have been investigated have not been shown to be effective. Currently, the mainstay of care for patients with ALS is timely intervention to manage symptoms, including use of nasogastric feeding, prevention of aspiration (control of salivary secretions and use of cough-assist devices), and provision of ventilatory support (usually with bilevel positive airway pressure). Some interventions raise serious ethical issues, such as whether to perform tracheostomy for full ventilation and, if so, when and how to withdraw respiratory support once it has been instituted.
Studies in cells, mice, and humans support the view that several types of reagents (e.g., antisense oligonucleotides and AAV-delivered microRNA) inactivate production of toxic gene products and thus may be therapeutic in ALS mediated by genes such as SOD184-87 and C9ORF72. Indeed, clinical trials investigating the use of antisense oligonucleotides to silence SOD1 have begun.
One can anticipate continued progress in understanding the biology of ALS. There is no doubt that high-throughput genetics, combined with improved clinical phenotyping, will further refine the genetic landscape of ALS. As thousands of full genome sequences become available, it will be feasible to explore the possibility that complex interactions among multiple gene variants explain not only familial ALS but also sporadic ALS. The exploration of environmental factors in sporadic ALS will expand, with a focus on the internal environment represented by the microbiome. The ultimate proof of our understanding of the biology of ALS will hinge on our ability to modify the clinical course of the disease (Brown and Al-Chalabi 2017).
Riluzole was the first FDA approved treatment for ALS, and, although the mechanism of action is poorly understood, is speculated to reduce glutamatergic neurotransmission, by blocking voltage-gated sodium channels on presynaptic neurons. In the original trial, Riluzole, increased 18-month survival of patients by 3 months compared with placebo, but had no significant effect on muscle strength. Riluzole is a relatively safe drug, although the most common adverse effects are an increase in liver enzymes and asthenia (that is, a lack of energy) and some cases of fatal hepatic failure and pancreatitis have been reported. In addition to the traditional tablet form of the drug, an oral suspension has been produced and marketed in some countries for patients who are unable to swallow solid forms of the drug, owing to severe dysphagia. Edaravone, which is thought to act as an anti-oxidant agent has a beneficial effect on progression in a highly selected cohort of patients with early onset and rapidly progressive disease, and accordingly, has been licensed by the US FDA but not by the European Medicines Agency. Whether Edaravone should be provided to all patients of ALS regardless of clinical presentation is a matter of debate (Hardiman and others, 2017).
Clinical trials
The Australian New Zealand Clinical Trials Registry lists clinical trials including ALS/MND studies in Australia and New Zealand, as well as trials from across the globe that have been completed, are currently recruiting or are pending. There are currently over 80 ALS/MND clinical trials being conducted worldwide. The Clinical Trial page on this website provides information on those being conduced in Australia. The ALS Signal: Clinical Research Dashboard developed by the organisation I Am ALS illustrates and lists clinical trials being undertaken globally.
Special Access Scheme
The Special Access Scheme (SAS) is managed by the Therapeutic Goods Administration (TGA). SAS refers to arrangements which provide for the import and/or supply of an unapproved therapeutic good for a single patient, on a case by case basis. SAS applications are made to the TGA by registered medical practitioners, preferably the treating doctor.
Off-label prescribing
Sometimes a medicine may be licensed for one condition, but could have the potential to be used to treat other conditions or illnesses. This is referred to as 'off-label' use of an 'unlicensed medicine'. Off-label prescribing is when a registered medicine is prescribed for use that is not included in the approved product information. An unlicensed or off-label medication may be prescribed by doctors if they think it is likely to be effective for their patient and any benefits outweigh potential side effects or risks.
ALS Untangled provides reviews by clinicians and scientists of alternative and off-label MND drugs.
The Australian MND Registry was established in 2004 as a means to facilitate the collection and analysis of MND patient data in Australia. The goal of the registry is to improve patient care through continuous evaluation of patient management and associated outcomes, and by contributing to research. Following registration of the patient by a neurologist, other providers such as the GP or nurse can update patient details. Data from the Australian MND Registry has recently been transferred to a new integrated registry developed through the MiNDAUS partnership as outlined below. The aim of this integrated approach is to improve care, inform policy and facilitate patient involvement in research.
The Australian MND registry is an observational cohort study initiated in 2004. This registry has collected a data set of, demographic, clinical profiles and disease progression data at a national level from 2005 to 2015. In Australia, this methodology was initially developed within a single comprehensive ALS clinic, and then expanded to a national level. The primary aim of the national registry was to help coordinate focused care and a foundation for case ascertainment for research in ALS/MND (Talman et al 2016).
The Sporadic ALS Australia Systems Genomics Consortium (SALSA-SGC) was established in 2015 funded by the MND Research Australia Ice Bucket Challenge Grant. The consortium is led by Professor Naomi Wray, from the University of Queensland and Associate Professor Ian Blair, from Macquarie University, NSW. SALSA collects consistent clinical data and biological samples across clinics in Australia to underpin future research based on biological samples, including genomics.
The MiNDAUS Partnership, was established in 2018 with funding from the National Health and Medical Research Council, Australian MND organisations and private donors The MiNDAUS Partnership is constructing a secure online platform with two distinct parts: The MiNDAUS Patient Registry, and the MiNDAUS Clinical Registry. From 2022 the MiNDAUS Clinical Registry has replaced the Australian Motor Neurone Disease Registry (AMNDR) and will offer linkage to the SALSA genomics project.
The MiNDAus Partnership builds on and extends existing national collaborations in a targeted approach to improve the standard and coordination of care for people living with MND in Australia, and to enhance the prospects of discovering a cure or treatment. Relationships have been developed between leading clinical and research groups as well as patient-centered organizations, care providers, and philanthropy with a shared vision. MiNDAus has established a corporate structure and meets at least biannually to decide on how best to progress research, drug development, and patient management. The key themes are; (i) empowering patients and their family carers to engage in self-management and ensure personalized service provision, treatment, and policy development, (ii) integration of data collection so as to better inform policy development, (iii) unifying patients and carers with advocacy groups, funding bodies, clinicians and academic institutions so as to inform policy development and research, (iv) coordination of research efforts and development of standardized national infrastructure for conducting innovative clinical MND trials that can be harmonized within Australia and with international trials consortia (Vucic et al 2021).
Motor Neurone Disease Research Australia is the research arm of MND Australia. MNDRA promotes, supports and funds MND research with the greatest chance of making an impact on the future of MND. The MND Australia Research Committee reviews grant applications and determines the distribution of funds within set policies, and according to stringent scientific assessment criteria.
Information on current research, projects funded by the MNDRA and clinical trials is available in the research section of this website.
Project MinE is an international groundbreaking genetic ALS research initiative established to understand the genetic basis of ALS and to ultimately find a cure. Project MinE aims to analyse the DNA of at least 15,000 ALS patients and 7,500 controls. The resulting 22,500 DNA profiles will be compared. To date 10,904 DNA profiles have been collected and many new genetic mutations have been discovered.
Shoesmith et al 2021, Canadian best practice recommendations for the management of amyotrophic lateral sclerosis:
See - disease-modifying therapies and medication alignment recommendations - table 1
Andersen et al 2012, EFNS guidelines on the Clinical Management of Amyotrophic Lateral Sclerosis (MALS) – revised report of an EFNS task force, Neuroprotective treatment/disease-modifying treatment recomendations:
- Patients with ALS should be offered treatment with riluzole 50 mg twice daily (level A).
- Treatment should be initiated as early as possible after diagnosis (GCPP). Realistic expectations for treatment effects and potential side effects should be discussed with the patient and caregivers (GCPP).
- Patients with progressive muscular atrophy, primary lateral sclerosis or hereditary spastic paraplegia should as a rule not be treated with riluzole (GCPP).
- Irrespective of familial disposition, all patients with a symptomatic progressive MND and carrying a SOD1 gene mutation should be offered treatment with riluzole (GCPP).
- Currently, there is insufficient evidence to recommend treatment with vitamins, testosterone, antioxidants such as co-enzyme Q-10 and gingko biloba, intravenous immunoglobulin therapy, cyclosporin, interferons, Copaxone, KDI tripeptide, neurotrophic factors (including BDNF, IGF-1 and mecasermin rinfabate), ceftriaxone, creatine, gabapentin, minocycline, stem cells or lithium (GCPP).
Miller et al 2009, Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: drug, nutritional, and respiratory therapies (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology.
See also: Summary of Evidence Based Guideline for Clinicians
National Institute for Health and Clinical Excellence (2001) NICE Guideline: Guidance on the use of Riluzole (Rilutek) for the treatment of Motor Neurone Disease
MND Aware e-training program MND Hub – online training course for health professionals and service providers developed by MND NSW - Session 1: What is MND?
MND Research Australia, State of Play - a webinar series profiling MND research developments in Australia.
International Alliance of ALS/MND Associations Scientific Advisory Council (SAC), Briefing notes on clinical trials of potential therapies
International Alliance of ALS/MND Association, webinar series 2021:
- Biomarkers - discussion chaired by Dr David Taylor, ALS Canada
- Optimizing Clinical Trial Design - discussion chaired by Dr Gethin Thomas, MND Australia
- Introduction to ALS/MND Genetics - Dr Kelly Williams and Professor Ammar Al Chalabi
- Genetics Counselling and Testing - Dr. Adriano Chiò, Dr Alisdair McNeill, Jennifer Roggenbuck
MND Association of England, Wales and Northern Ireland, Research Blog
MND Association of England, Wales and Northern Ireland, Podcast series, MND Matters Episode 5: Research
ALS TDI Endpoints podcast series, The Endpoints Podcast features guests from the ALS community, including people living with ALS, researchers, advocates and fundraisers. Each episode cuts to the chase with short, digestible conversations about the latest research at ALS TDI and the most closely watched ALS clinical trials as well as stories from people living with ALS with their thoughts on research, healthcare and their journey with ALS.
Stem Cells Australia, information on stem cells and motor neurone disease
International ALS/MND Connect, Perth, 2019, research updates, Professor Matthew Kiernan "Hope is on the horizon: Advances in research", Professor Ammar Al-Chalabi "Causes of ALS/MND", and Professor Leonard van den Berg "Clinical trials- accelerating ALS/MND therapy development through innovative designs".
International Alliance of ALS/MND Associations Annual meeting 2018:
- Scientific update, Dr David Taylor, ALS Canada.
International Alliance of ALS/MND Associations,15th Allied Professionals Forum 2017:
Resources to download
More About MND: A guide for people living with MND, their family and friends
Fact sheets
Cognitive and behavior change in MND
Familial MND and genetic testing
Useful web pages:
About MNDState MND Associations
Other resources
MND Decision Assist Tool, ‘Should I have predictive genetic testing for motor neurone disease?
ALS TDI- ALS Town Hall series:
healthtalk.org - stories collected by academic researchers who interview people in their own homes, using their own words:
MND NSW, Ask the Experts 2020:
- The Genetics of MND: What does it mean to me?, Rosie Fell and Ashley Crook (at 12.10 minutes)
MND Australia, 4th MND Connect, 2018:
- The MND jigsaw: patterns within the puzzle, Dr Thanuja Dharmadasa
- Genes and the environment: different causes of MND, Ken Rogers
- The role of stem cells in screening drugs as potential therapies, Associate Professor Bradley Turner
MND Australia, 2nd MND Connect 2016:
- The Australian MND Registry, Associate Professor Paul Talman